Abstract
Background Ethnic differences have the potential to confound associations between HIV-1 subtype and immunologic progression. We compared declines in CD4 cell counts during untreated infection for the most prevalent HIV-1 subtypes, focusing on distinguishing between the effects of viral subtype and ethnicity.
Methods We combined data from 4 European and 6 Canadian cohorts, selecting adults in the stable chronic phase of untreated HIV infection. We estimated the change in square root CD4 cell count over time for subtypes and ethnicities using mixed models, adjusting for covariates selected for their potential effect on initial CD4 cell count or its decline.
Results Data from 9772 patients were analyzed, contributing 79 175 measurements of CD4 cell count and 24 157 person-years of follow-up. Overall, there were no appreciable differences in CD4 cell count decline for viral subtypes A, CRF01_AE, CRF02_AG, C and G compared with viral subtype B; whereas the decline in CD4 cell count in patients of African ancestry was considerably slower than in patients of other ethnicity. When ethnic groups were studied separately, there was evidence for slower declines in CD4 cell count in viral subtypes C, and possibly A and G, compared with viral subtype B in patients of African ancestry but not among patients of other ethnicities, suggesting an interaction between subtype and ethnicity.
Interpretation Ethnicity is a major determinant of CD4 cell count decline; viral subtype differences may have existed but were small compared with the effect of ethnicity and were most apparent in patients of African ancestry. In developing countries, slower CD4 cell count declines among individuals of African descent may translate to a longer asymptomatic phase and increase the opportunity for HIV transmission.
HIV-1 has acquired extensive genetic diversity with 9 recognized subtypes (A−D, F−H, J and K) and many circulating recombinant forms (CRFs).1,2 Subtype B is the most studied because it is the predominant virus in North America and Europe. Most (89%) HIV infections worldwide, however, originate from non−subtype-B viruses, with subtypes C and A and recombinants CRF01_AE and CRF02_AG accounting for 73% of all HIV-1 infections.3 Owing to patterns of immigration, a growing number of HIV infections in developed countries are occurring in migrant communities with HIV infection resulting from viral subtypes other than subtype B,4,5 with increasing transmission of subtypes between ethnicities.6 In Canada, the other subtypes now account for about 15% of newly reported HIV infections.7
CD4 cell counts are both the primary prognostic marker for HIV-related disease and the main indicator for starting antiretroviral therapy. Worldwide use of antiretroviral therapy is expanding, and it is important to understand whether differences in immunologic progression exist that might necessitate subtype-specific monitoring and treatment guidelines. Subtype-specific estimates of CD4 cell count trajectories are also essential for public health models used to predict the course of the HIV epidemic and are helpful for estimating what proportion of persons born abroad acquire HIV infection in their adopted country.8
HIV-1 diversity might affect immunologic progression through differential interactions with the human host.9–13 Determining subtype-specific effects on immunologic progression is challenging:14 non−subtype-B viruses predominantly affect individuals of African descent, and several studies have suggested that rates of decline in the CD4 cell count differ according to ethnicity. Ethnic differences have the potential to confound any association between viral subtype and immunologic progression.15–17 We conducted a Canadian and European collaborative study to examine CD4 cell count decline, according to the most prevalent viral subtypes, in untreated patients who are in the stable chronic phase of HIV infection, and we focused on distinguishing between the effects of viral subtype and ethnicity.
Methods
Data collection
Ten cohorts or cohort collaborations were selected: 4 from Europe and 6 from Canada (Table 1).18–23 The project was approved by the relevant scientific boards or steering committees. EuroSIDA omitted patients also enrolled in other contributing cohorts. For each patient, we requested all available CD4 cell counts in patients enrolled in the cohort until the start of antiretroviral therapy, and the dates of any newly acquired AIDS-defining illness or death.
Seven cohorts provided information on ethnicity using a variety of classifications including race categories (e.g., black, white, Asian and Aboriginal), country of origin or a combination of the two (Table 1); therefore, ethnicity was coded as African ancestry, other ethnicity or unknown. French law does not permit the ANRS CO3 Aquitaine cohort to collect information on ethnicity or country of origin. Viral subtype was determined by the REGA HIV-1 and HIV-2 automated subtyping tool (version 2.0) based on HIV-1 pol sequences during genotypic resistance testing or, in some earlier cases, with the Subtype Analyzer tool.24
Our analysis was restricted to adults (age ≥ 16 yr) enrolled in the studies between January 1998 and June 2010 with a known HIV-1 subtype and at least 2 CD4 cell count measurements within 1 year while they were naive to antiretroviral therapy. We selected CD4 cell count measurements in patients who were within a stable chronic phase of untreated HIV infection. Various strategies have been used in studies of CD4 cell count decline to exclude measurements made in the acute or late phases of infection.16,17,25,26 In this study, we omitted any patient with a first AIDS event either before or within 3 months after their first CD4 cell count measurement; these patients were likely to be late presenters. To avoid including CD4 cell count measurements made during acute infection, we removed measurements within 6 months of documented seroconversion (known only for 15% of patients) and measurements < 100 cells/μL at the beginning of a series; these measurements may reflect the rapid CD4 cell count decline that occurs immediately after seroconversion before the immune system responds to infection. To avoid including measurements made in late-stage infection, we ended a patient’s series when the first of 2 consecutive measurements fell below 100 cells/μL. Consecutive measurements at both the beginning and end of the series had to be at least 2 months and no more than 12 months apart to avoid remeasurement (where a second measurement was made to check a first) or measurement prompted by clinical deterioration (where a patient lost to follow-up returned because of poor health).
Statistical analysis
Patients were followed from the time of the first CD4 cell count measurement in their series until the start of antiretroviral therapy, the last CD4 cell count measurement in their series or death. In the main analysis, we compared CD4 cell counts (square root transformed to stabilize the variance) over time for the most common (at least 100 patients available) HIV-1 subtypes (A, CRF01_AE, CRF02_AG, B, C and G) using a mixed linear regression model. This mixed model included both patient-specific random intercepts and random slopes over time to allow for heterogeneity in patients’ initial CD4 cell counts and subsequent declines. In the main analysis, models were adjusted for covariates selected because of their potential effect on initial CD4 cell count (age, sex, ethnicity, initial HIV RNA, injection drug use as the likely mode of transmission and cohort) or on CD4 cell count decline (age, sex, ethnicity, initial HIV RNA and injection drug use as the likely mode of transmission). In a descriptive analysis of ethnic and subtype effects, we plotted random effects representing each patient’s estimated CD4 cell count decline from a mixed model without ethnicity or subtype slope parameters. We also report CD4 cell count intercept and slope estimates for each subtype from mixed models fit to untransformed data, because rates of CD4 cell count decline are easier to interpret on the original scale than on a square root scale.
We explored the results from our main analysis in a variety of sensitivity analyses. First, the main analysis model was refit to all CD4 cell counts since January 1998 without further selection (to include measurements made in both the acute and advanced stages of infection). Second, separate mixed models were fit for patients of African and other ethnicities. Third, the main analysis model was fit as part of a joint model, together with an exponential model for the time taken to start antiretroviral therapy.27 The joint model allows for informative censoring if the CD4 cell count series are shorter for some subtypes because patients with these subtypes start therapy at higher CD4 cell counts. Finally, we varied the covariates and cohorts in the analysis because not all cohorts were able to provide all covariates of interest.
Clinical events, such as time to a first AIDS-defining illness and death before starting antiretroviral therapy, were summarized according to viral subtype and ethnicity. No formal analyses were conducted because of the small numbers of events and the potential for bias due to unobserved events before cohort enrolment.
We used SAS 9.2 (SAS Institute, Cary, North Carolina) for all analyses.
Results
A total of 14 092 patients met the inclusion criteria: 13 682 patients had one of the subtypes A, CRF01_AE, CRF02_AG, B, C or G (Figure 1). After applying CD4 cell count selection criteria, 9772 patients remained, contributing 79 175 CD4 cell count measurements and 24 157 person-years of follow-up with a median of 6 CD4 cell count measurements per patient (interquartile range [IQR] 3−11). For most patients, CD4 cell count measurements were censored either by the patient starting antiretroviral therapy (37%) or by irregular measurement at the end of the series (33%), but 144 patients (1.5%) reached a CD4 cell count of < 100 cells/μL without starting antiretroviral therapy, and 22 patients (0.2%) died.

Selection of study cohort. *At least 100 patients available per subtype with CD4 cell counts within the stable chronic phase of untreated infection. †Includes patients with ethnicity assigned based on country of origin, n = 246 (2.5%) (Table 1).
Subtype B accounted for most of the HIV-1 infections (81.2%) followed by subtypes C (8.4%), A (3.6%), CRF02_AG (3.6%), CRF01_AE (2.0%) and G (1.1%) (Table 2). The demographic and clinical characteristics of the other viral subtypes were similar, with the exception of CRF01_AE. Non−subtype-B infections were found principally among patients of African ancestry, whereas subtype B and CRF01_AE infections were found among those of other ancestries. Both initial CD4 cell counts and initial HIV RNA were higher in patients with HIV-1 infections caused by subtypes CRF01_AE or B.
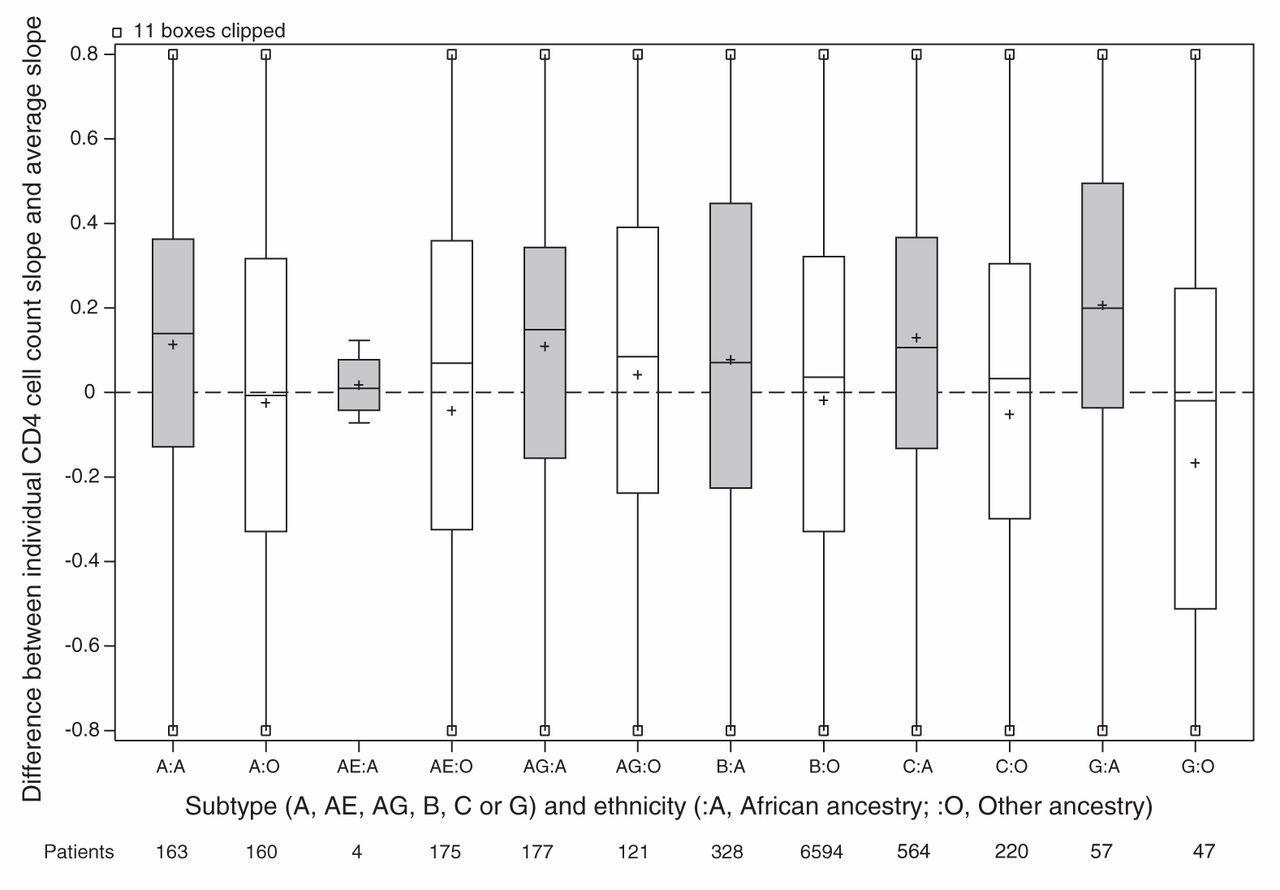
For the main analysis, we omitted 1162 patients because either their ethnicity or initial HIV RNA was unknown (Figure 1). Patients of unknown ethnicity had characteristics similar to those of other ethnicities (Table 3). In the main analysis, patients of African ancestry had an appreciably slower rate of CD4 cell count decline compared with patients of other ethnicities (Table 4). Differences between viral subtypes, however, were minor compared with the difference between the 2 ethnic categories. Subtype differences became apparent when the main analysis model was fit to unselected CD4 cell counts, with appreciably slower declines in cell counts in patients with viral subtypes A, C and CRF02_AG compared with patients with viral subtype B. If ethnicity was ignored in an analysis of selected CD4 cell counts, then these 3 subtypes appeared to have slower declines than patients with viral subtype B. In our descriptive analysis of ethnicity and viral subtype effects, patients of African ancestry had slower declines among all subtypes compared with patients of other ethnicities (Figure 2), including within subtype B (although no conclusion can be drawn about subtype CRF01_AE). In an exploratory analysis of patients with subtype B, patients of African ancestry from Caribbean countries appeared to have a CD4 decline intermediate between other patients of African ancestry and patients of other ethnicities (Figure 3).
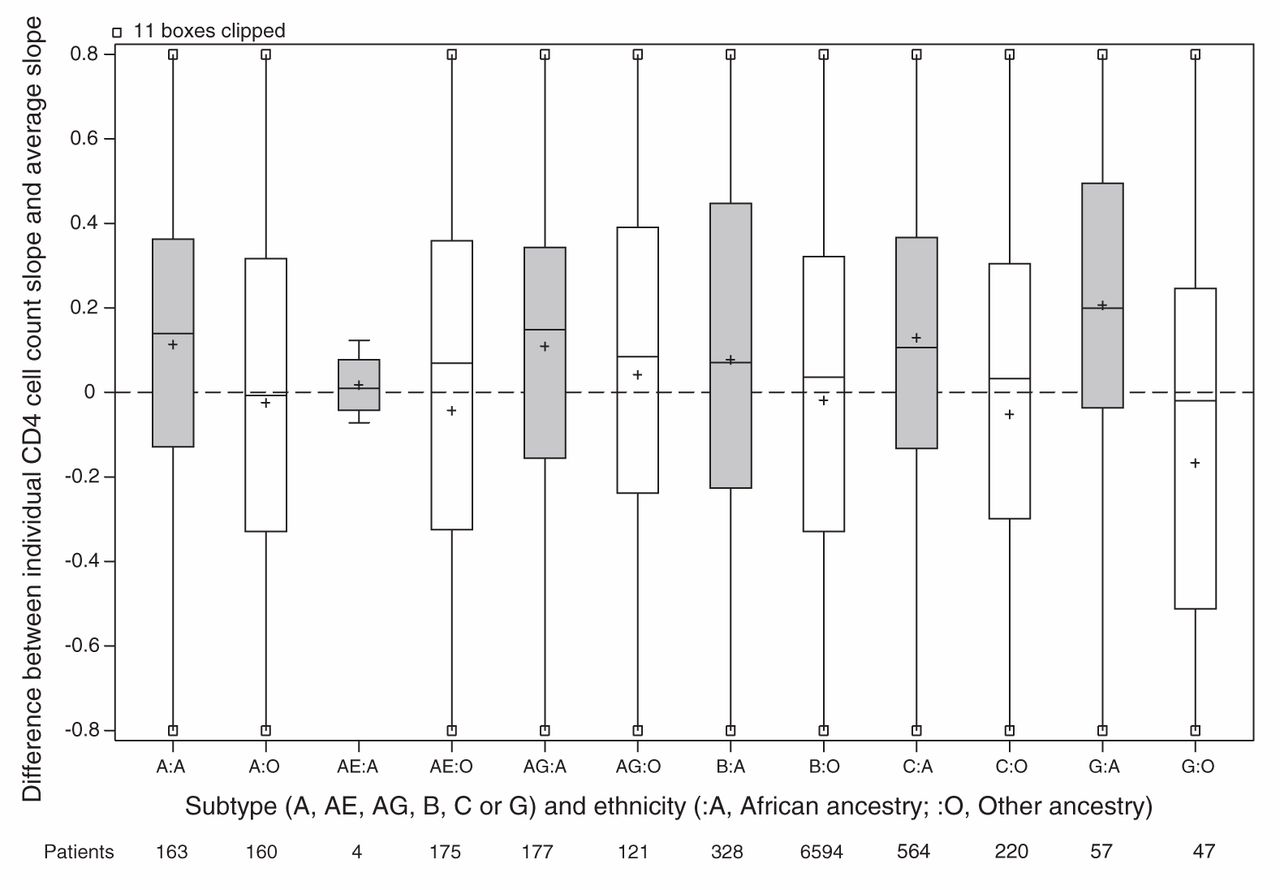
Estimated CD4 cell count slope decline for each patient. Random effects representing each patient’s CD4 cell count decline (compared with the average) estimated in a mixed model without ethnicity or subtype slope parameters but adjusted for covariates. All cohorts are included in the model except Aquitaine. Grey shading = patients of African ancestry, white shading = patients of other ethnicities, + = mean.
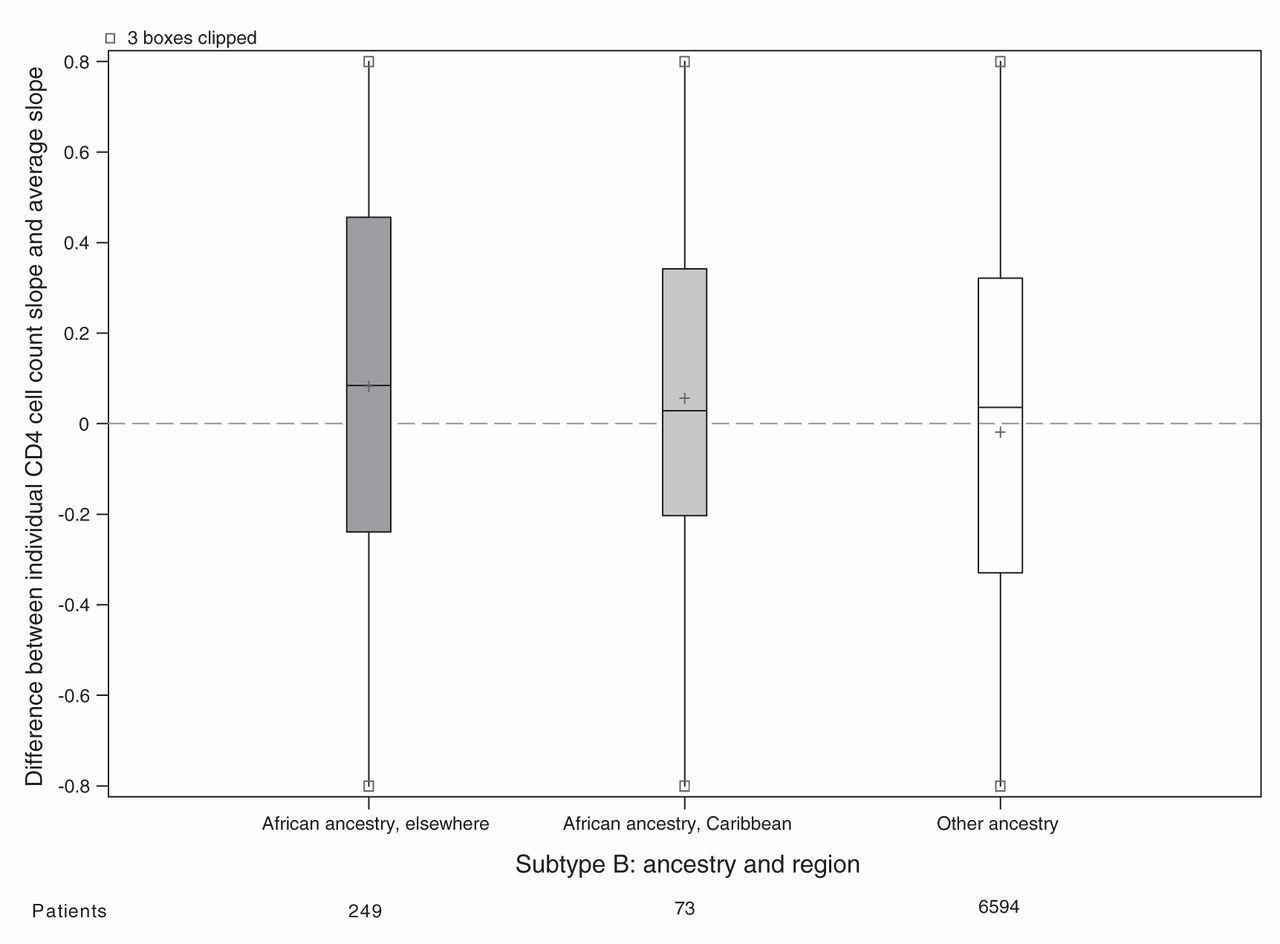
Estimated decline in CD4 cell count slope for each patient with viral subtype B. Random effects represnting each patient’s CD4 cell count decline (compared with the average) estimated in a mixed model without ethnicity or subtype slope parameters but adjusted for covariates. All cohorts are represented in the model except Aquitaine; however, the only patients shown in this plot have HIV subtype B. Grey shading = patients of African ancestry, white shading = patients of other ethnicities, + = mean.
In sensitivity analyses, we fit separate models for patients of African and other ethnicities (Table 4). The results from these analyses suggest little difference between subtypes in patients of other ethnicities, but patients of African ancestry with viral subtype C and possibly subtypes A and G showed slower declines compared with viral subtype B. Only viral subtype CRF01_AG appeared to have a similar effect in both ethnic groups. This amounts to an interaction between the effects of subtype and ethnicity; however, we did not have sufficient data to estimate this interaction in a single model. Our data suggest that there is little variation between viral subtypes in both initial CD4 cell count and its decline in patients of other ethnicities (Table 5). Model parameters imply that it would take a reference patient of African ancestry with viral subtype C 1.2 years to reach a threshold of 350 cells/µL and 9.3 years to reach a threshold of 200 cells/µL, whereas a reference patient of another ethnicity with viral subtype B would take 2.5 years to reach a threshold of 350 cells/µL and 6.2 years to reach a threshold of 200 cells/µL (footnote to Table 5). Figure 4 shows the estimated average CD4 cell count decline over 5 years in patients of African ancestry with viral subtype C and in patients of other ethnicities with viral subtype B.
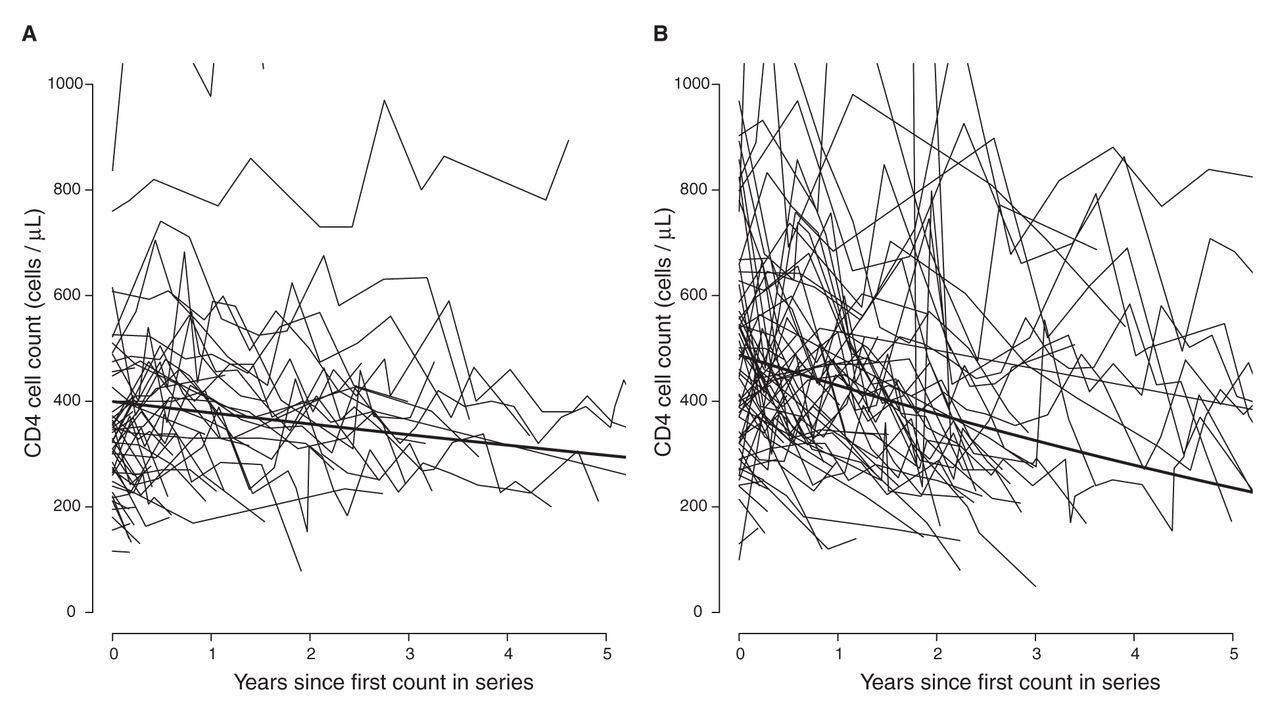
Estimated average CD4 cell count over 5 years. The average CD4 cell count estimated (A) in patients of African ancestry with viral subtype C and (B) in patients of other ethnicities with viral subtype B. Estimates were made using separate mixed models: one for patients of African ancestry and one for patients of other ethnicities and represent CD4 cell counts in a male patient with infection not transmitted by injection drug use and with age and initial HIV RNA set at the median for that ethnicity and subtype. CD4 cell count trajectories are shown for a 10% sample of patients of African ancestry with viral subtype C and for a 1% sample of patients of other ethnicities with viral subtype B.
Estimates of CD4 cell count decline did not change appreciably in a joint model (Table 6). In the longitudinal component of the joint model, lower initial CD4 cell count and more rapid decline were associated with older age, female sex, injection drug use as the likely mode of transmission and higher initial HIV RNA. In the time-to-event component of the joint model, earlier antiretroviral therapy was associated with older age, female sex, higher initial HIV RNA, more recent calendar time and African ancestry. There was no evidence from this model that viral subtype and ethnicity slope estimates were biased by informative censoring because of early antiretroviral therapy.
In other sensitivity analyses, we added a time-dependent CD8 cell count (not available from the EuroSIDA cohort); we replaced injection drug use as the likely mode of transmission with covariates for hepatitis C and chronic hepatitis B virus co-infection (not available from the HAART Observational Medical Evaluation and Research Study (HOMER) cohort). Subtype and ethnicity slope estimates were not materially different in these analyses.
We report clinical outcomes using unselected CD4 cell count series (Table 7) because selected series were often censored by irregular measurement. There were no appreciable differences between viral subtypes and ethnicities in either the proportion of patients starting antiretroviral therapy or the median CD4 cell count when starting. However, the median time taken to start antiretroviral therapy was shorter among patients of African ancestry (0.51 v. 1.27 yr; Table 7), consistent with their lower initial CD4 cell count (400 v. 470 cells/µL; Table 3). Within each viral subtype, no more than 14% of patients had an AIDS-defining illness, and 1% of patients died during a median follow-up at around 2 years. There was no obvious relationship between the number of these clinical events and the rate of CD4 cell count decline by either viral subtype or ethnicity.
Interpretation
Main findings
The study explores the effects of both viral subtype and ethnicity on CD4 cell count decline in untreated patients during the chronic phase of HIV infection.14 We focused on the chronic phase because it is the longest period of untreated HIV infection. We were able to compare all of the predominant subtypes directly with one another. Indeed, the proportions of viral subtypes studied (other than B) mirrored their proportions in the worldwide HIV epidemic.3 We found that ethnicity is the major determinant of CD4 cell count decline; viral subtype differences may exist but were small compared with the effect of ethnicity and were most apparent in patients of African ancestry. Patients of African ancestry had slower rates of CD4 cell count decline overall. Although the viral subtypes other than B, with the exception of CRF01_AE, appeared to be associated with slower CD4 cell count decline in models that did not include a slope for African ancestry, these effects were greatly attenuated once this slope was included. Furthermore, in plots of CD4 cell count decline by viral subtype and ethnicity, patients of African ancestry had slower rates of CD4 cell count decline within each viral subtype, including within viral subtype B. Together these results suggest that differences between viral subtypes are small compared with the broader effect of ethnicity on immunologic progression. Failure to account for ethnicity in the evaluation of viral subtype effects could potentially lead to erroneous conclusions.
Strong associations between viral subtype and ethnicity exist. Only 5% of patients with viral subtype B and 3% of those with CRF01_AE infection were of African ancestry compared with more than 50% of those infected with other subtypes. We therefore examined the effects of viral subtype in each ethnic group separately. Among patients of other ethnicities, there was little evidence for an effect of viral subtype on immunologic progression. In contrast, patients of African ancestry with viral subtype C (and possibly A, CRF02_AG and G) may have slower rates of decline in cell count compared with patients of African ancestry with viral subtype B. This suggests viral subtype may have different effects depending on ethnicity.
Explanation and comparisons with other studies
Indeed, biological differences between viral subtypes do exist that might affect immunologic progression. For example, coreceptor use varies among viral subtypes, with A and C predominantly using the coreceptor (R5) that is associated with slower disease progression, and viral subtype C has lower replicative fitness when compared with other viral subtypes in vitro.13 Observational studies also support differences in disease progression among patients of African ancestry with infections of differing viral subtype. For example, faster immunologic progression has been reported in patients from Uganda and Kenya with viral subtype D compared with viral subtype A.28–30 Co-evolution of HIV-1 subtypes and African populations may reduce the virulence of more dominant viral subtypes, such as subtype C, possibly leading to an interaction between viral subtype and ethnicity.2 In the absence of antiretroviral therapy, which is still the reality for most people infected with HIV in Africa, our results suggest that a patient of African ancestry with a non−subtype-B infection could take considerably longer to reach a CD4 cell count of 200 cells/µL (9.3 v. 6.2 yr; Table 5), the level at which the risk of AIDS increases, thus prolonging the asymptomatic period and increasing the opportunity for HIV transmission.31,32 Because of the small number of clinical events and deaths, we were not able to determine if differences in CD4 cell count decline would translate into different risks of AIDS or mortality for people of different ethnicities.
Several previous studies in both incident33,34 and prevalent4,16,17 cohorts have shown that African ancestry is associated with slower rates of CD4 cell count decline, but with one exception,33 have not included viral subtype and ethnicity in the same analysis.14 Yet, it remains unclear how ethnicity could affect the underlying pathogenesis of HIV infection.35 Those patients classified as being of African ancestry in our study came from a large number of African and Caribbean countries, suggesting no single host characteristic is likely to be responsible for differences in disease progression. Differential patterns of migration and racial admixture in Caribbean populations should reduce differences owing to ethnicity; this was shown in an analysis where patients from Haiti and patients from Canada (both sets of patients with subtype B infection) had similar rates of CD4 cell count decline, which were faster than the rate observed among recent immigrants from Africa.36 In our study, patients with subtype B infection from the Caribbean appeared to have rates of CD4 cell count decline intermediate to rates of CD4 cell count decline in patients with subtype B infection from Africa and other countries.
Individuals of similar ethnicity do share genetic characteristics that have been associated with HIV disease progression. Examples include protective human leukocyte antigen haplotypes, chemokine and chemokine receptor polymorphisms, and mutational variants in genes involved in immune regulation.37,38 It is also possible that ethnicity simply serves as a marker for socioeconomic, cultural and environmental factors that may influence immunologic progression.31 In our study, patients of African ancestry were more likely to be female and of child-bearing age, have lower initial CD4 cell counts and to have started antiretroviral therapy sooner. However, our joint model showed no evidence of informative censoring owing to early antiretroviral therapy. The healthy migrant effect is an alternate explanation for differences between patients of African ancestry and other ethnicities receiving health care in developed countries,39 but this does not explain reported differences in patients from Africa or the United States.15,16,34,40 In our study, adjustment for time-updated CD8 cell count, a crude marker of immune activation, or for hepatitis co-infection did not affect estimates of differences in CD4 cell count decline. A healthy migrant effect implies that ethnic differences should be attenuated after adjusting for comorbidities.
Limitations
Our classification of ethnicity was rudimentary based on different classifications in each cohort or, for 2 cohorts, derived from country of origin, and ethnicity was unknown for 11% of patients. We also had insufficient patients of Asian ancestry to pursue a separate study. Patients included in this study were undergoing treatment in countries with publicly funded health care, minimizing the potential effects of health care access and quality on immunologic progression, but our results might not be generalizable to resource-limited settings. We did not have enough patients with other viral subtypes linked to faster CD4 cell count decline, such as viral subtype D.28–30,40 In addition, limited numbers of both viral subtypes G and CRF01_AE in patients of African ancestry means our estimates for these subtypes were imprecise. We were not able to adjust for sociodemographic factors beyond age and sex. Multiple-host, socioeconomic and environmental factors are shared among individuals of common ancestry, which may impact CD4 cell count decline. Single-cohort studies are needed to see if ethnic differences within the same viral subtype are attenuated with adjustment for specific socioeconomic and environmental factors.
Conclusions and implications for practice and future research
This large collaborative analysis with a broad representation of the most common HIV-1 subtypes worldwide suggests that ethnicity is more prognostic of immunologic progression than viral subtype during untreated chronic HIV infection. Although there is some evidence of differences in CD4 cell count decline between viral subtypes, particularly in patients of African ancestry, research to uncover the underlying biologic or sociologic reasons for slower immunologic progression among patients of African ancestry is warranted. Further study of subtype-specific effects on immunologic progression will require comparisons of different subtypes in ethnically homogeneous populations.
Supplemental information
For reviewer comments and the original submission of this manuscript, please see www.cmajopen.ca/content/2/4/E318/suppl/DC1
Acknowledgements
The authors thank the patients and researchers who contributed to this study: Montreal Chest Institute Chronic Viral Illness Cohort, Montréal, Que.; HAART Observational Medical Evaluation and Research (HOMER) study, Vancouver, B.C. (http://www.cfenet.ubc.ca/research/homer/team); Southern Alberta Clinic Cohort, Calgary, Alta.; Toronto General Hospital, Toronto, Ont.; Ottawa General Hospital, Ottawa, Ont.; McMaster University, Hamilton, Ont.; The UK Collaborative HIV Cohort (UK CHIC) (http:/212.219.75.232/UKCHIC/StudyOrganisationUKCHIC.asp); the UK HIV Drug Resistance Database (www.hivrdb.org.uk); Swiss HIV Cohort Study (www.shcs.ch/26-organisation-shcs); the multi-centre study group on EuroSIDA (www.cphiv.dk/Ongoing-Studies/EuroSIDA/Study-group); the Groupe d’Epidémiologie Clinique du Sida en Aquitaine (GECSA) − ANRS CO3 (http://gecsa.isped.u-bordeaux2.fr/Presentation.aspx). In particular, the authors wish to acknowledge the contributions of Bernard Masquelier (Centre Hospitalier Universitaire (CHU), Hôpital Pellegrin, Laboratoire de Virologie, Bordeaux, France) who provided virologic data for the ANRS CO3 Aquitaine cohort and participated in revising earlier drafts of the manuscript. Bernard passed away in March 2013. His collegiality and contributions to the field of HIV resistance will be greatly missed.
Footnotes
-
Competing interests: Marina Klein received grants from Merck, the Canadian Institutes of Health Research (CIHR), the National Institute of Health Research, Fonds de recherche du Québec − Santé and Schering-Plough, consulting fees from ViiV, and lecture fees from Bristol-Meyers Squibb and ViiV. She also received fees for the development of educational presentations from Gilead and ViiV. Caroline Sabin received grants from the Medical Research Council of England and Wales during the conduct of the study. Darrell Tan received grants from Gilead and ViiV, consulting fees from Gilead and ViiV, and lecture fees from Abbott, Bristol-Myers Squibb, Gilead, Janssen, Merck and ViiV. Sharon Walmsley received grants, consulting fees, lecture fees, nonfinancial support and fees for the development of educational presentations from Merck, ViiV, Gilead, Abbott, Tibotec, Janssen, , Bristol-Myers Squibb and Boehringer Ingelheim. John Gill received a grant from the CIHR and personal fees for being a member of the national advisory boards of Abbvie, Gilead, Merck, Janssen, ViiV and Bristol-Myers Squibb. No competing interests were declared by the other authors.
-
Contributors: Marina Klein had full access to the data and takes responsibility for the integrity of the data and the accuracy of the reported findings. Jim Young had full access to the dataset, conducted all data analysis and acts as guarantor for the analyses. All of the authors participated in discussions on the design of the study and interpretation of the findings, revised the manuscript, approved the final version submitted for publication and agreed to act as guarantors of the work.
-
Funding: This work was supported by Fonds de recherche du Québec − Santé–Réseau SIDA/maladies infectieuses for the Montreal Chest Immunodeficiency cohort. Marina Klein is supported by a Chercheur-nationaux career award from Fonds de recherche du Québec − Santé. The Swiss HIV Cohort Study is supported by the Swiss National Science Foundation (33CS30_134277). The UK Collaborative HIV Cohort Study Study and UK HIV Drug Resistance Database are funded by a program grant from the Medical Research Council UK (G06000337); the views expressed in this manuscript are those of the researchers and not necessarily those of the Medical Research Council. Primary support for EuroSIDA is provided by the European Commission BIOMED 1 (CT94-1637), BIOMED 2 (CT97-2713) and by the 5th Framework (QLK2-2000-00773), the 6th Framework (LSHP-CT-2006-018632), and the 7th Framework (FP7/2007-2013, EuroCoord No. 260694) programs. Current support for EuroSIDA also includes unrestricted grants by Gilead, Pfizer and Merck. The ANRS CO3 Aquitaine Cohort is funded in part by the French Agence Nationale de Recherches sur le Sida et les hépatites virales, together with the French Institut National de la Santé et de la Recherche Médicale and the Centre Hospitalier Universitaire de Bordeaux (COREVIH Aquitaine and CIC-EC 7). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
References
- © 2014 Canadian Medical Association or its licensors
In this issue

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Article tools